16. September 2020
Document
Design of Oral FXIIIa Blockers as Safer Anticoagulants Mission Impossible?
by Martin Stieler (September 2020)
Thromboembolic events such as heart attack, stroke and pulmonary embolism are among the leading causes of death in industrialized countries. The common strategy to reduce the risk of blood vessel obstruction (thrombosis) is to administer substances reducing the coagulation process (anticoagulants). However, all current anticoagulants like heparins, cumarins (Phenprocoumon/Marcumar®, Warfarin) as well as the latest direct-acting oral anticoagulants (Dabigatran/Pradaxa®, Rivaroxaban/Xarelto®, Apixaban/Eliquis®, Edoxaban/Lixiana®) interfere substantially with the blood coagulation system resulting in a considerably elevated life-threatening bleeding risk.1 Consequently, there is still an unmet medical need for safer anticoagulants with a broad therapeutic window.
For decades, blood coagulation factor XIII (FXIII) is discussed as a promising target for the development of safer drugs with reduced bleeding risk compared to current anticoagulants.2,3 However, no compound blocking FXIIIa entered clinical trials yet. In the last years, knowledge especially in structural biology and medicinal chemistry in the scope of FXIII and other human transglutaminases increased substantially. Thus, achieving the goal to develop oral-available FXIII inhibitors for safer antithrombotic therapy appears feasible for the first time.
Unique feature of FXIIIa blocker: No impairment of thrombin level
FXIII is the last enzyme of the blood coagulation cascade and cross-links fibrin fibers of the so called “soft” clot (Figure 1). In addition, FXIII decorates the blood clot with α2-antiplasmin preventing premature degradation by plasmin. Both cross-linking and α2-antiplasmin incorporation increase the biochemical and physicochemical stability of the mature clot. Consequently, FXIIIa inhibition reduces the clot stiffness rendering FXIII as a target for antithrombotic treatment.
The outstanding role of FXIII in the coagulation cascade is that it acts downstream of thrombin. Thus, inhibition of FXIIIa would not affect the level of thrombin and all the other coagulation factors. In consequence, neither platelet activation nor fibrin aggregation resulting in the formation of a “soft” clot are disturbed. As a result, the bleeding risk supposed to be potentially lower compared to current anticoagulants.
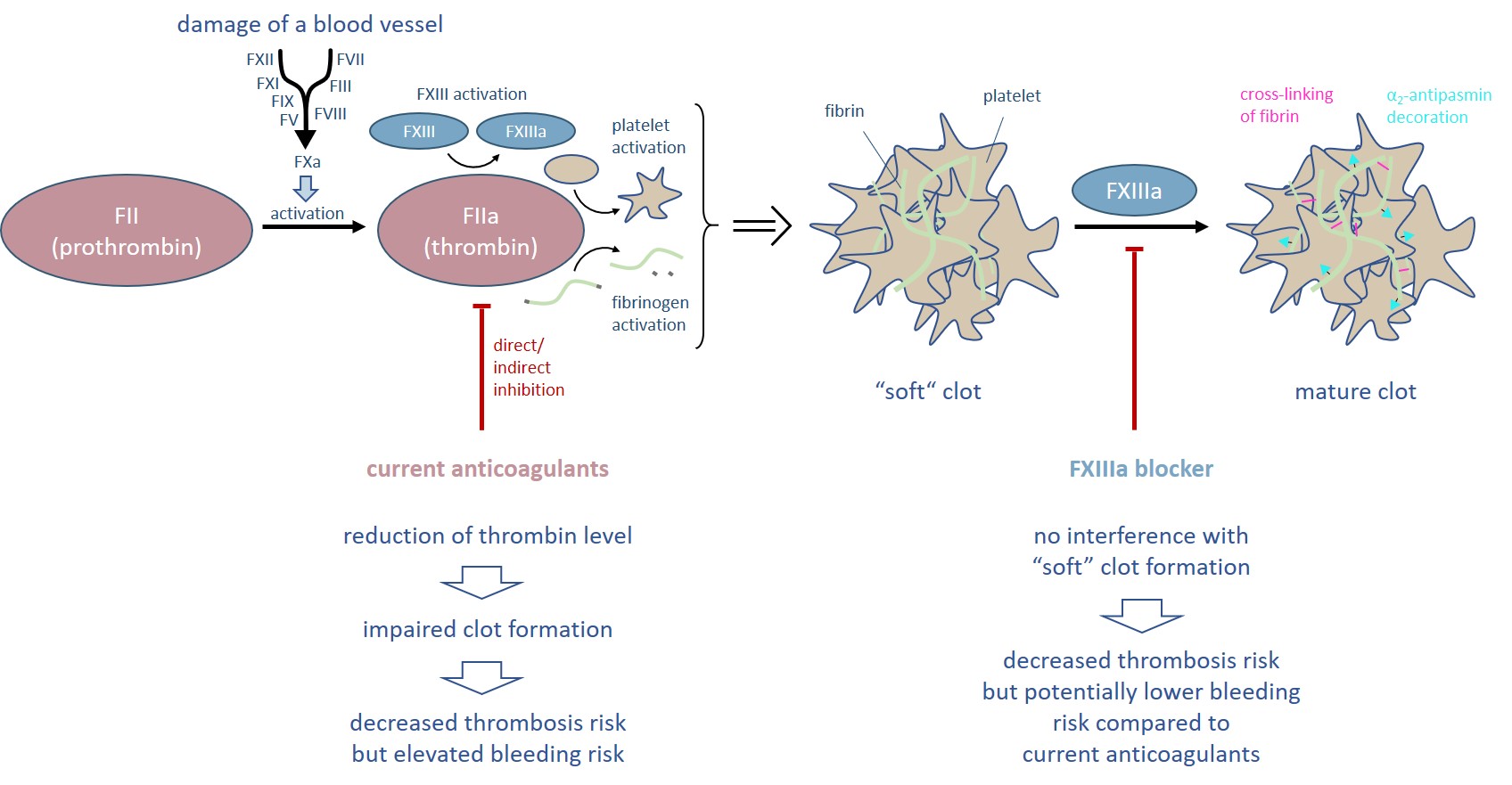
Figure 1. Thrombin is the central enzyme of the blood coagulation process and initiates the formation of a “soft” clot. All current anticoagulants directly or indirectly reduce the thrombin level and thus are associated with an elevated bleeding risk. In contrast, inhibition of FXIIIa allows the formation of a “soft” clot.
In 2019, the Zedira team has published the first drug-like direct-acting FXIIIa blocker.4 “Rotational thromboelastometry indicated that the inhibitor ZED3197 (Figure 2), in a dosedependent manner, slightly prolonged clot formation, reduced clot firmness and facilitated clot lysis without affecting the clotting time, indicating minimal impact on hemostasis. Furthermore, in a mechanistic animal model (rabbit), the novel FXIIIa inhibitor effectively decreased the weight of clots and facilitated flow restoration without prolongation of the bleeding time. In conclusion, both thromboelastometry and the in vivo data suggest an anticoagulative effect without increased bleeding tendency.”
These promising features of ZED3197 qualify the compound as drug candidate for short term anticoagulation especially in the acute care setting. Zedira is looking for partnering the project to promote this compound into clinical development.
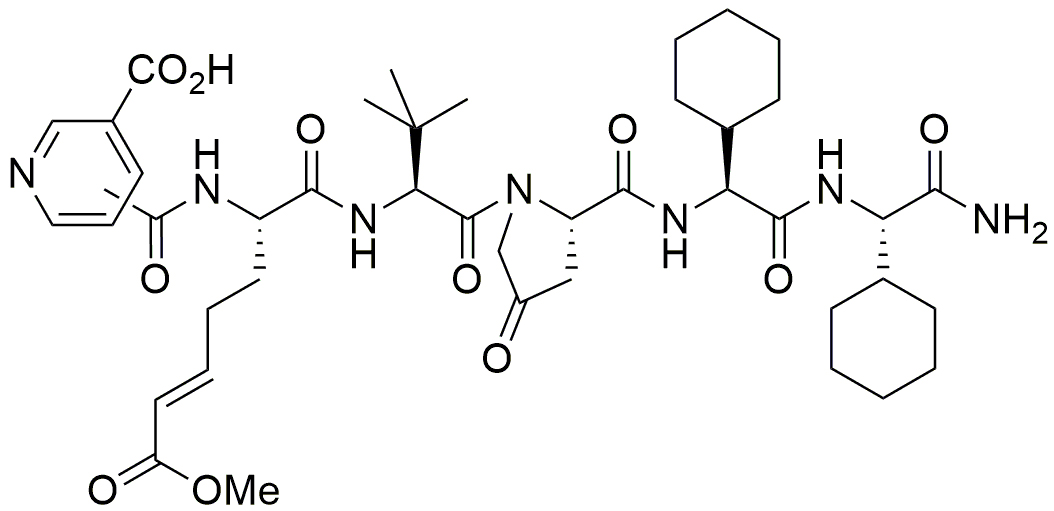
Figure 2. The inhibitor ZED3197 consists of six artificial amino acids. The reactive glutamine is substituted by an α,β-unsaturated carbonic acid ester (Michael-acceptor) that binds covalently to Cys314 of the catalytic triad.
A more detailed blog article considering the potential of FXIIIa as molecular target for safer anticoagulants was posted by Alisa Wolberg in 2015: Factor XIIIa: novel target for anticoagulation?
The crystal structures of FXIII in the active state brings light into darkness
In 2013, we have published the crystal structure of FXIII in the active state (FXIIIa) revealing a remarkable conformational change during substrate binding (Figure 3).5 Both β-barrel domains flip aside (red arrow) exposing the active site. Remarkably, in the active state three calcium ions are coordinated affecting the shape of the active site of FXIIIa. The crystal structure provides a detailed map at atomic level deciphering the interaction of our lead compound ZED1301 with the target.
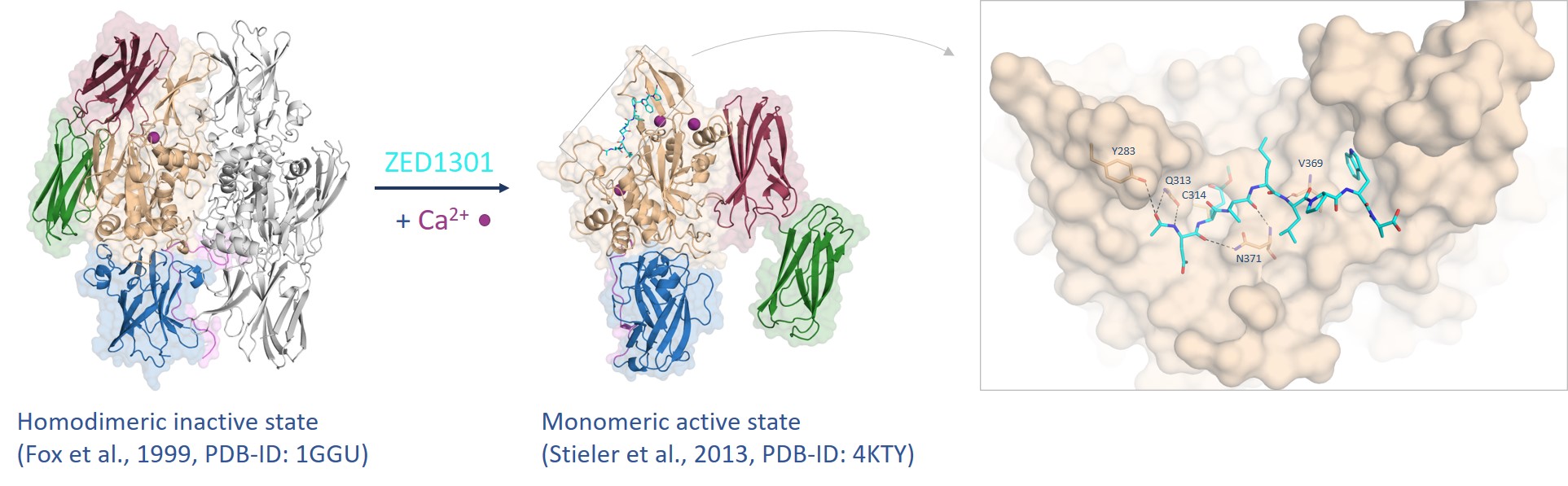
Figure 3. FXIII undergoes a tremendous conformational change from the inactive to the active state assisted by calcium coordination (purple spheres) whereas the inhibitor ZED1301 (cyan) binds tightly to the surface-exposed and shallow active site.5,6
The inhibitor ZED1301 bears (analogous to ZED3197) an α,β-unsaturated carbonic acid ester forming a covalent bond to the nucleophilic Cys314 of the catalytic triad. Such irreversible acting inhibitors are characterized by high potency and are widely used in drugs like Aspirin® or Penicillin®.7 Nevertheless, our armamentarium in FXIIIa-targeting should include reversible-acting compounds especially for the long-term treatment of certain risk patients. For this purpose, the inhibitors need to be reduced in their size, must become more rigid, less peptidic and less polar – but potency and selectivity must be preserved.
Due to the extensive conformational change, it can be hypothesized that the active state has higher energy compared to the inactive state. Consequently, the inhibitor needs to be more potent than an inhibitor that binds to a conventional target adopting the same conformation in the bound and unbound state. The first available crystal structure of FXIIIa in complex with ZED1301 (Figure 3) shows that the binding site is exposed to the surface and does not bear deep pockets enabling the development of reversible inhibitors with high affinity.
In conclusion, the challenging “mission” in terms of reversible FXIII inhibition is that the extensive conformational change requires over-average potent inhibitors. However, the development of high-affinity inhibitors is hampered by the fact that the binding site is located at the surface and has a shallow shape.
Within a lead design project a deep hydrophobic pocket was revealed additional to the already established hydrophobic pocket (Figure 4) enabling the synthesis of more potent inhibitors.8 Herein, it was shown that starting from a methyl thiazole (ZED1630) the transient hydrophobic pocket can be occupied by the introduction of an ethyl ester (ZED2360) resulting in a significant increase in affinity. This study shows that the active site of FXIIIa is less shallow than originally expected and the synthesis of more potent inhibitors is viable by occupation of the novel deep hydrophobic pocket.
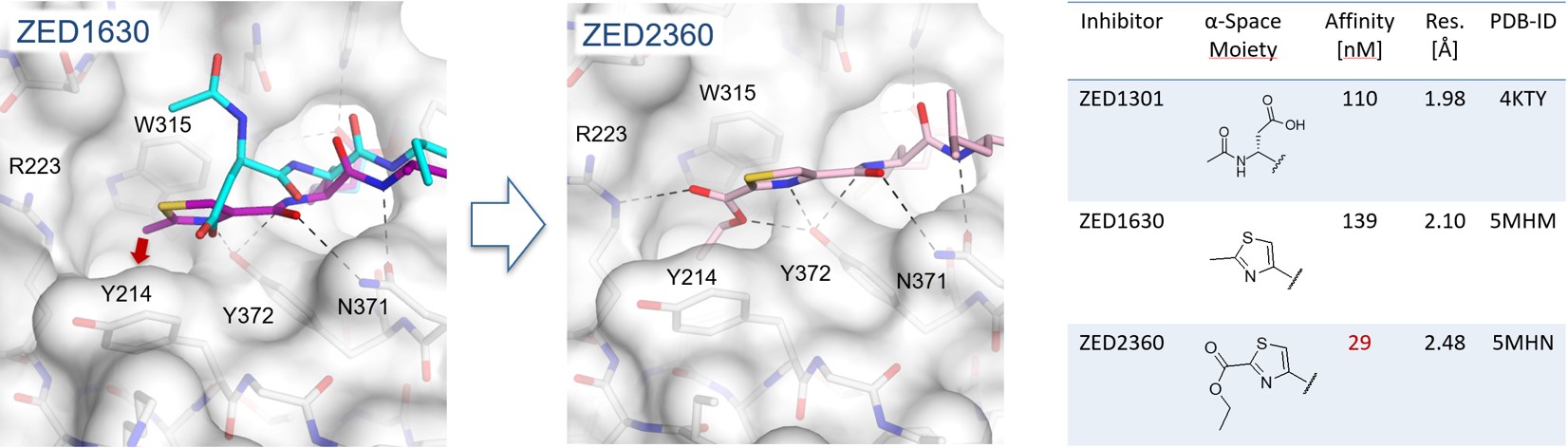
Figure 4. In the crystal structure of FXIIIa in complex with ZED1630 (purple) a novel deep hydrophobic pocket has been revealed (red arrow). The methyl thiazole binds in contrast to the structural aligned ZED1301 (cyan) closer to the protein surface (grey). Addressing the hydrophobic pocket by the ethyl ester of ZED2360 (pink) leads to a fivefold increase in binding affinity.
Drugs addressing TG2 pave the way for oral FXIIIa blockers
With ZED1227 (Figure 5), a TG2 blocker for celiac disease treatment, the first compound targeting an enzyme of the transglutaminase family is investigated in clinical trials (EudraCT Number: 2017-004807-31). Depending on the clinical data, transglutaminases might be validated as druggable targets for the first time (see press release: Dr. Falk Pharma and Zedira announce start of the phase 2a proof of concept study of ZED1227 for the treatment of celiac disease)
Figure 5. The peptide-mimetic inhibitor ZED1227, clinical drug candidate for celiac disease, bears an α,β-unsaturated carbonic acid ester (Michael-acceptor) analogous to ZED1301. The compound targets active tissue transglutaminase (TG2a) in the inflamed digestive system to treat the gluten-induced autoimmune disease.
In addition, Zedira recently completed a project dealing with the development of reversibly acting TG2 inhibitors for the treatment of fibrotic disorders. Within this project, the company developed novel molecules with an extraordinary binding affinity bearing an α-ketoamide instead of the Michael-acceptor warhead (see press release: Reversibly acting transglutaminase 2 inhibitors: drug candidates for the treatment of fibrosis).
ZED3197 is the first drug-like compound for potential antithrombotic therapy addressing FXIIIa. The physicochemical features of the inhibitor predispose for i.v. application in the acute care setting. Moreover, the promising in vivo results motivate the Zedira team, despite of the challenging aspects as discussed in this article, to work on a FXIII blocker for oral administration.
Under consideration of the rising knowledge concerning the structural biology of FXIII along with further insights gained by drug discovery projects targeting tissue transglutaminase, we are convinced that the design of “oral FXIII inhibitors” for safer antithrombotic therapy is a possible mission. We “just” must combine the already existing knowledge of our successfully completed projects.
References:
The author:

Thromboembolic events such as heart attack, stroke and pulmonary embolism are among the leading causes of death in industrialized countries. The common strategy to reduce the risk of blood vessel obstruction (thrombosis) is to administer substances reducing the coagulation process (anticoagulants). However, all current anticoagulants like heparins, cumarins (Phenprocoumon/Marcumar®, Warfarin) as well as the latest direct-acting oral anticoagulants (Dabigatran/Pradaxa®, Rivaroxaban/Xarelto®, Apixaban/Eliquis®, Edoxaban/Lixiana®) interfere substantially with the blood coagulation system resulting in a considerably elevated life-threatening bleeding risk.1 Consequently, there is still an unmet medical need for safer anticoagulants with a broad therapeutic window.
For decades, blood coagulation factor XIII (FXIII) is discussed as a promising target for the development of safer drugs with reduced bleeding risk compared to current anticoagulants.2,3 However, no compound blocking FXIIIa entered clinical trials yet. In the last years, knowledge especially in structural biology and medicinal chemistry in the scope of FXIII and other human transglutaminases increased substantially. Thus, achieving the goal to develop oral-available FXIII inhibitors for safer antithrombotic therapy appears feasible for the first time.
Unique feature of FXIIIa blocker: No impairment of thrombin level
FXIII is the last enzyme of the blood coagulation cascade and cross-links fibrin fibers of the so called “soft” clot (Figure 1). In addition, FXIII decorates the blood clot with α2-antiplasmin preventing premature degradation by plasmin. Both cross-linking and α2-antiplasmin incorporation increase the biochemical and physicochemical stability of the mature clot. Consequently, FXIIIa inhibition reduces the clot stiffness rendering FXIII as a target for antithrombotic treatment.
The outstanding role of FXIII in the coagulation cascade is that it acts downstream of thrombin. Thus, inhibition of FXIIIa would not affect the level of thrombin and all the other coagulation factors. In consequence, neither platelet activation nor fibrin aggregation resulting in the formation of a “soft” clot are disturbed. As a result, the bleeding risk supposed to be potentially lower compared to current anticoagulants.
Figure 1. Thrombin is the central enzyme of the blood coagulation process and initiates the formation of a “soft” clot. All current anticoagulants directly or indirectly reduce the thrombin level and thus are associated with an elevated bleeding risk. In contrast, inhibition of FXIIIa allows the formation of a “soft” clot.
In 2019, the Zedira team has published the first drug-like direct-acting FXIIIa blocker.4 “Rotational thromboelastometry indicated that the inhibitor ZED3197 (Figure 2), in a dosedependent manner, slightly prolonged clot formation, reduced clot firmness and facilitated clot lysis without affecting the clotting time, indicating minimal impact on hemostasis. Furthermore, in a mechanistic animal model (rabbit), the novel FXIIIa inhibitor effectively decreased the weight of clots and facilitated flow restoration without prolongation of the bleeding time. In conclusion, both thromboelastometry and the in vivo data suggest an anticoagulative effect without increased bleeding tendency.”
These promising features of ZED3197 qualify the compound as drug candidate for short term anticoagulation especially in the acute care setting. Zedira is looking for partnering the project to promote this compound into clinical development.
Figure 2. The inhibitor ZED3197 consists of six artificial amino acids. The reactive glutamine is substituted by an α,β-unsaturated carbonic acid ester (Michael-acceptor) that binds covalently to Cys314 of the catalytic triad.
A more detailed blog article considering the potential of FXIIIa as molecular target for safer anticoagulants was posted by Alisa Wolberg in 2015: Factor XIIIa: novel target for anticoagulation?
The crystal structures of FXIII in the active state brings light into darkness
In 2013, we have published the crystal structure of FXIII in the active state (FXIIIa) revealing a remarkable conformational change during substrate binding (Figure 3).5 Both β-barrel domains flip aside (red arrow) exposing the active site. Remarkably, in the active state three calcium ions are coordinated affecting the shape of the active site of FXIIIa. The crystal structure provides a detailed map at atomic level deciphering the interaction of our lead compound ZED1301 with the target.
Figure 3. FXIII undergoes a tremendous conformational change from the inactive to the active state assisted by calcium coordination (purple spheres) whereas the inhibitor ZED1301 (cyan) binds tightly to the surface-exposed and shallow active site.5,6
The inhibitor ZED1301 bears (analogous to ZED3197) an α,β-unsaturated carbonic acid ester forming a covalent bond to the nucleophilic Cys314 of the catalytic triad. Such irreversible acting inhibitors are characterized by high potency and are widely used in drugs like Aspirin® or Penicillin®.7 Nevertheless, our armamentarium in FXIIIa-targeting should include reversible-acting compounds especially for the long-term treatment of certain risk patients. For this purpose, the inhibitors need to be reduced in their size, must become more rigid, less peptidic and less polar – but potency and selectivity must be preserved.
Due to the extensive conformational change, it can be hypothesized that the active state has higher energy compared to the inactive state. Consequently, the inhibitor needs to be more potent than an inhibitor that binds to a conventional target adopting the same conformation in the bound and unbound state. The first available crystal structure of FXIIIa in complex with ZED1301 (Figure 3) shows that the binding site is exposed to the surface and does not bear deep pockets enabling the development of reversible inhibitors with high affinity.
In conclusion, the challenging “mission” in terms of reversible FXIII inhibition is that the extensive conformational change requires over-average potent inhibitors. However, the development of high-affinity inhibitors is hampered by the fact that the binding site is located at the surface and has a shallow shape.
Within a lead design project a deep hydrophobic pocket was revealed additional to the already established hydrophobic pocket (Figure 4) enabling the synthesis of more potent inhibitors.8 Herein, it was shown that starting from a methyl thiazole (ZED1630) the transient hydrophobic pocket can be occupied by the introduction of an ethyl ester (ZED2360) resulting in a significant increase in affinity. This study shows that the active site of FXIIIa is less shallow than originally expected and the synthesis of more potent inhibitors is viable by occupation of the novel deep hydrophobic pocket.
Figure 4. In the crystal structure of FXIIIa in complex with ZED1630 (purple) a novel deep hydrophobic pocket has been revealed (red arrow). The methyl thiazole binds in contrast to the structural aligned ZED1301 (cyan) closer to the protein surface (grey). Addressing the hydrophobic pocket by the ethyl ester of ZED2360 (pink) leads to a fivefold increase in binding affinity.
Drugs addressing TG2 pave the way for oral FXIIIa blockers
With ZED1227 (Figure 5), a TG2 blocker for celiac disease treatment, the first compound targeting an enzyme of the transglutaminase family is investigated in clinical trials (EudraCT Number: 2017-004807-31). Depending on the clinical data, transglutaminases might be validated as druggable targets for the first time (see press release: Dr. Falk Pharma and Zedira announce start of the phase 2a proof of concept study of ZED1227 for the treatment of celiac disease)
Figure 5. The peptide-mimetic inhibitor ZED1227, clinical drug candidate for celiac disease, bears an α,β-unsaturated carbonic acid ester (Michael-acceptor) analogous to ZED1301. The compound targets active tissue transglutaminase (TG2a) in the inflamed digestive system to treat the gluten-induced autoimmune disease.
In addition, Zedira recently completed a project dealing with the development of reversibly acting TG2 inhibitors for the treatment of fibrotic disorders. Within this project, the company developed novel molecules with an extraordinary binding affinity bearing an α-ketoamide instead of the Michael-acceptor warhead (see press release: Reversibly acting transglutaminase 2 inhibitors: drug candidates for the treatment of fibrosis).
ZED3197 is the first drug-like compound for potential antithrombotic therapy addressing FXIIIa. The physicochemical features of the inhibitor predispose for i.v. application in the acute care setting. Moreover, the promising in vivo results motivate the Zedira team, despite of the challenging aspects as discussed in this article, to work on a FXIII blocker for oral administration.
Under consideration of the rising knowledge concerning the structural biology of FXIII along with further insights gained by drug discovery projects targeting tissue transglutaminase, we are convinced that the design of “oral FXIII inhibitors” for safer antithrombotic therapy is a possible mission. We “just” must combine the already existing knowledge of our successfully completed projects.
References:
- M. Shoeb, M. C. Fang, Assessing bleeding risk in patients taking anticoagulants JTH 35, 312-319 (2013).
- L. Lorand, A. Jacobsen, Accelerated Lysis of Blood Clots. Nature 195, 91-912 (1962).
- R. J. Shebuski, G. R. Sitko, D. A. Claremon, J. J. Baldwin, D. C. Remy, A. M. Stern, Inhibition of Factor-XIIIa in a Canine Model of Coronary-Thrombosis - Effect on Reperfusion and Acute Reocclusion after Recombinant Tissue-Type Plasminogen-Activator. Blood 75, 1455-1459 (1990).
- R. Pasternack, C. Büchold, Robert Jähnig, Christiane Pelzer, Michael Sommer, Andreas Heil, Peter Florian, Götz Nowak, Uwe Gerlach, M. Hils, Novel inhibitor ZED3197 as potential drug candidate in anticoagulation targeting coagulation FXIIIa (F13a). JTH 18, 191-200 (2020).
- M. Stieler, J. Weber, M. Hils, P. Kolb, A. Heine, C. Büchold, R. Pasternack, G. Klebe, Structure of Active Coagulation Factor XIII Triggered by Calcium Binding: Basis for the Design of Next-Generation Anticoagulants. Angew Chem Int Edit 52, 11930-11934 (2013).
- B. A. Fox, V. C. Yee, L. C. Pedersen, I. Le Trong, P. D. Bishop', R. E. Stenkamp, D. C. Teller, Identification of the calcium binding site and a novel ytterbium site in blood coagulation factor XIII by X-ray crystallography. J Biol Chem 274, 4917-4923 (1999).
- J. Singh, R. C. Petter, T. A. Baillie, A. Whitty, The resurgence of covalent drugs. Nat Rev Drug Discov 10, 307-317 (2011).
- M. Stieler, C. Büchold, M. Schmitt, A. Heine, M. Hils, R. Pasternack, G. Klebe, StructureBased Design of FXIIIaBlockers: Addressing a Transient Hydrophobic Pocket in the Active Site of FXIIIa. ChemMedChem 15, 900-905 (2020).
The author:
Martin Stieler studied chemistry at the University of Marburg and has a PhD in Medicinal Chemistry. His expertise comprises protein crystallography and rational drug design with focus on transglutaminases. Currently, he leads the department of structural biology at Zedira GmbH.
Document
 Successful ISO9001:2015 recertification
Successful ISO9001:2015 recertification  Besuch des Bundesministers für Wirtschaft und Klimaschutz Dr. Robert Habeck bei der Zedira
Besuch des Bundesministers für Wirtschaft und Klimaschutz Dr. Robert Habeck bei der Zedira  Discover Our New Catalogue Edition and Dive into the World of Transglutaminases!
Discover Our New Catalogue Edition and Dive into the World of Transglutaminases!  Successful ISO9001:2015 recertification
Successful ISO9001:2015 recertification  Dr. Falk Pharma and Zedira announce successful completion of the phase 2a proof-of-concept study of ZED1227 for the treatment of Celiac Disease
Dr. Falk Pharma and Zedira announce successful completion of the phase 2a proof-of-concept study of ZED1227 for the treatment of Celiac Disease  Dr. Falk Pharma und Zedira verkünden den erfolgreichen Abschluss der Phase 2a-Studie mit ZED1227 zur Behandlung von Zöliakie
Dr. Falk Pharma und Zedira verkünden den erfolgreichen Abschluss der Phase 2a-Studie mit ZED1227 zur Behandlung von Zöliakie  Reversibly acting transglutaminase 2 inhibitors: drug candidates for the treatment of fibrosis
Reversibly acting transglutaminase 2 inhibitors: drug candidates for the treatment of fibrosis  Transcriptomic analysis of the efficacy of TG2-inhibitor trials and human intestinal organoids modelling Celiac disease pathogenesis
Transcriptomic analysis of the efficacy of TG2-inhibitor trials and human intestinal organoids modelling Celiac disease pathogenesis  Transglutaminase antibodies and neurological manifestations of gluten sensitivity
Transglutaminase antibodies and neurological manifestations of gluten sensitivity  Design of Oral FXIIIa Blockers as Safer Anticoagulants Mission Impossible?
Design of Oral FXIIIa Blockers as Safer Anticoagulants Mission Impossible?  Microbial transglutaminase (MTG) enables efficient and site-specific conjugation to native antibodies without the need of antibody engineering
Microbial transglutaminase (MTG) enables efficient and site-specific conjugation to native antibodies without the need of antibody engineering  Tridegin as FXIIIa inhibitor
Tridegin as FXIIIa inhibitor  Microbial transglutaminase: from discovery to market
Microbial transglutaminase: from discovery to market  Tissue transglutaminase inhibitors
Tissue transglutaminase inhibitors  Tissue transglutaminase in Alzheimers Disease
Tissue transglutaminase in Alzheimers Disease  Factor XIIIa: novel target for anticoagulation?
Factor XIIIa: novel target for anticoagulation?  Microbial transglutaminase for site-specific protein conjugation
Microbial transglutaminase for site-specific protein conjugation