6. April 2016
Tissue transglutaminase inhibitors
by Jeffrey W. Keillor (University of Ottawa, Ottawa, Canada)
Tissue transglutaminase (TG2) is a complex, bifunctional enzyme that was first identified for its calcium-dependent ability to catalyze transamidation reactions of peptide-bound glutamine residues with primary amines, including peptide-bound lysine residues, which results in the cross-linking of proteins. In addition to this activity, TG2 is known to function as a G-protein that participates in receptor signalling inside the cell. These activities are strictly regulated by mechanisms that include a dramatic conformational change between an ‘open’ form that catalyzes acyl-transfer and a ‘closed’ form that binds guanine nucleotides.1
Reversible inhibitors
Our initial studies of TG2 were focussed on its ability to catalyze acyl transfer reactions,2 in particular the formation of secondary amides from a substrate as inactive as a primary amide – a remarkable feat of which very few enzymes are capable. During our investigation into the structural basis of the unusual affinity of the small molecule substrate Cbz-Gln-Gly, we made a serendipitous discovery: A synthetic impurity in the sample of a cinnamoyl substrate analogue acted as a potent, reversible inhibitor of the transamidation reaction. This led us to prepare several series of cinnamoyl inhibitors3 (Figure 1), including CP4d, which was shown to be a competitive inhibitor with respect to the acyl-donor substrate (Ki = 174 nM).4 Photolabelling studies5 showed these inhibitors are bound in the acyl-donor substrate binding site, and more recent conformational6 and kinetic studies7 suggest they may bind more strongly to the closed conformation of TG2.
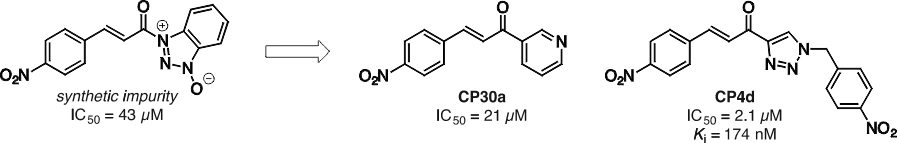 Figure 1. Synthetic impurity (left), serendipitously discovered to be an inhibitor of TG2, that led to the development of more potent reversible inhibitors (right).
Figure 1. Synthetic impurity (left), serendipitously discovered to be an inhibitor of TG2, that led to the development of more potent reversible inhibitors (right).
Irreversible inhibitors
Several research groups have made outstanding contributions to the development of irreversible inhibitors of TG2, some of which show promising biological activity (see reviews).8,9,10,11 Following an approach commonly employed in the field, we have designed several series of irreversible inhibitors of TG2 by decorating a variety of scaffolds with a variety of electrophilic warheads. Initially we explored inhibitors bearing acrylamide,12 epoxide,12 1,2,4-thiadiazole13 and maleimide14 warheads on the Cbz-X-Gly scaffold, as well as inhibitors armed with acrylamide, chloromethyl ketone and maleimide15 warheads on the Cbz-Phe-X scaffold. Subsequent acrylamide inhibitors based on more extended scaffolds have been used as probes in cells16,17,18 and tissues.19
More recently we have focussed on the conformational effects of inhibition. While some reversible inhibitors appear to favour the closed conformation of TG2,6,7 we have shown that irreversible inhibitors such as NC9 (Figure 2) appear to ‘lock’ the enzyme in its open conformation.6,20 Molecular modelling suggests this may be due to the inhibitor occupying certain binding pockets on the surface of the open conformation, which prevents protein folding into the compact conformation (Figure 3). In support of this hypothesis, inhibitors like NC9 have also been found to abolish GTP binding activity,21 presumably through an allosteric model whereby they lock TG2 in a conformation in which its GTP binding site is closed (Figure 4).
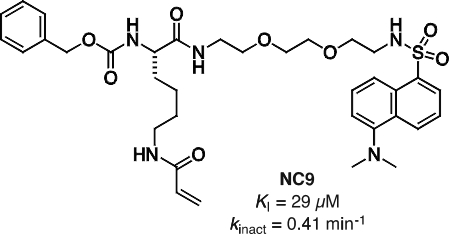
Figure 2. Structure and kinetic parameters of irreversible inhibitor NC9.
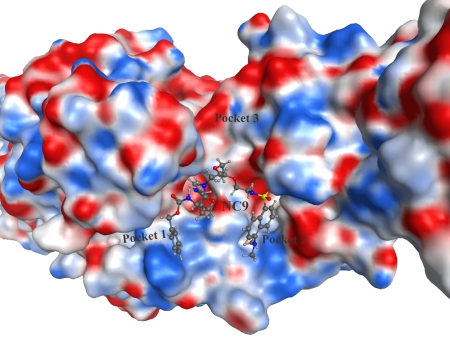
Figure 3. Irreversible inhibitor NC9 docked into the active site of ‘open’ TG2 (PDB code 2Q3Z22), showing relative position of binding pockets on the surface of the enzyme.
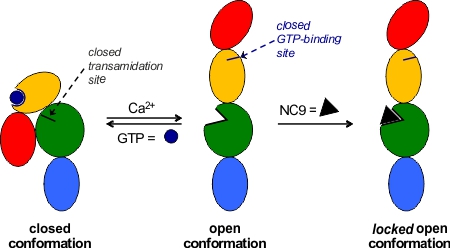
Figure 4. Allosteric model of irreversible inhibition of both transamidation and GTP-binding activity.
Our most recent collaborative work with Richard Eckert (Maryland) has demonstrated the biological relevance of this activity. The GTP-binding function of TG2 is essential for the survival of cancer stem cells, and pharmacological intervention with NC9 results in reduced survival, spheroid formation and migration.23,24 Further investigation is underway with Gail Johnson (Rochester) to study the effect of such inhibitors on glioblastoma multiforme (GBM). In the meantime, we have initiated structure-activity relationship (SAR) studies aimed at understanding the structural features that confer anti-cancer activity, in NC9 and more potent derivatives.
It is an honour to provide an overview of our work, as requested, for this blog. Furthermore, it is also an honour to be part of a community of medicinal chemists and biologists whose work inspires us to find more effective pharmacological solutions to TG2-related pathologies.
References:
The author:

Tissue transglutaminase (TG2) is a complex, bifunctional enzyme that was first identified for its calcium-dependent ability to catalyze transamidation reactions of peptide-bound glutamine residues with primary amines, including peptide-bound lysine residues, which results in the cross-linking of proteins. In addition to this activity, TG2 is known to function as a G-protein that participates in receptor signalling inside the cell. These activities are strictly regulated by mechanisms that include a dramatic conformational change between an ‘open’ form that catalyzes acyl-transfer and a ‘closed’ form that binds guanine nucleotides.1
Reversible inhibitors
Our initial studies of TG2 were focussed on its ability to catalyze acyl transfer reactions,2 in particular the formation of secondary amides from a substrate as inactive as a primary amide – a remarkable feat of which very few enzymes are capable. During our investigation into the structural basis of the unusual affinity of the small molecule substrate Cbz-Gln-Gly, we made a serendipitous discovery: A synthetic impurity in the sample of a cinnamoyl substrate analogue acted as a potent, reversible inhibitor of the transamidation reaction. This led us to prepare several series of cinnamoyl inhibitors3 (Figure 1), including CP4d, which was shown to be a competitive inhibitor with respect to the acyl-donor substrate (Ki = 174 nM).4 Photolabelling studies5 showed these inhibitors are bound in the acyl-donor substrate binding site, and more recent conformational6 and kinetic studies7 suggest they may bind more strongly to the closed conformation of TG2.
Irreversible inhibitors
Several research groups have made outstanding contributions to the development of irreversible inhibitors of TG2, some of which show promising biological activity (see reviews).8,9,10,11 Following an approach commonly employed in the field, we have designed several series of irreversible inhibitors of TG2 by decorating a variety of scaffolds with a variety of electrophilic warheads. Initially we explored inhibitors bearing acrylamide,12 epoxide,12 1,2,4-thiadiazole13 and maleimide14 warheads on the Cbz-X-Gly scaffold, as well as inhibitors armed with acrylamide, chloromethyl ketone and maleimide15 warheads on the Cbz-Phe-X scaffold. Subsequent acrylamide inhibitors based on more extended scaffolds have been used as probes in cells16,17,18 and tissues.19
More recently we have focussed on the conformational effects of inhibition. While some reversible inhibitors appear to favour the closed conformation of TG2,6,7 we have shown that irreversible inhibitors such as NC9 (Figure 2) appear to ‘lock’ the enzyme in its open conformation.6,20 Molecular modelling suggests this may be due to the inhibitor occupying certain binding pockets on the surface of the open conformation, which prevents protein folding into the compact conformation (Figure 3). In support of this hypothesis, inhibitors like NC9 have also been found to abolish GTP binding activity,21 presumably through an allosteric model whereby they lock TG2 in a conformation in which its GTP binding site is closed (Figure 4).
Figure 2. Structure and kinetic parameters of irreversible inhibitor NC9.
Figure 3. Irreversible inhibitor NC9 docked into the active site of ‘open’ TG2 (PDB code 2Q3Z22), showing relative position of binding pockets on the surface of the enzyme.
Figure 4. Allosteric model of irreversible inhibition of both transamidation and GTP-binding activity.
Our most recent collaborative work with Richard Eckert (Maryland) has demonstrated the biological relevance of this activity. The GTP-binding function of TG2 is essential for the survival of cancer stem cells, and pharmacological intervention with NC9 results in reduced survival, spheroid formation and migration.23,24 Further investigation is underway with Gail Johnson (Rochester) to study the effect of such inhibitors on glioblastoma multiforme (GBM). In the meantime, we have initiated structure-activity relationship (SAR) studies aimed at understanding the structural features that confer anti-cancer activity, in NC9 and more potent derivatives.
It is an honour to provide an overview of our work, as requested, for this blog. Furthermore, it is also an honour to be part of a community of medicinal chemists and biologists whose work inspires us to find more effective pharmacological solutions to TG2-related pathologies.
References:
- Eckert, R. L.; Kaartinen, M. T.; Nurminskaya, M.; Belkin, A. M.; Colak, G.; Johnson, G. V. W.; Mehta, K. Physiol. Rev. 2014, 94, 383–417.
- Keillor, J. W.; Clouthier, C. M.; Apperley, K. Y. P.; Akbar, A.; Mulani, A. Bioorg. Chem. 2014, 57, 186–197.
- Pardin, C.; Pelletier, J. N.; Lubell, W. D.; Keillor, J. W. J. Org. Chem. 2008, 73, 5766–5775.
- Pardin, C.; Roy, I.; Lubell, W. D.; Keillor, J. W. Chem. Biol. Drug Des. 2008, 72, 189–196.
- Pardin, C.; Roy, I.; Chica, R. A.; Bonneil, E.; Thibault, P.; Lubell, W. D.; Pelletier, J. N.; Keillor, J. W. Biochemistry 2009, 48, 3346–3353.
- Caron, N. S.; Munsie, L. N.; Keillor, J. W.; Truant, R. PLoS ONE 2012, 7, e44159.
- Clouthier, C. M.; Keillor, J. W. unpublished.
- Keillor, J. W.; Chabot, N.; Roy, I.; Mulani, A.; Leogane, O.; Pardin, C. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 78, 415–447.
- Keillor, J. W.; Apperley, K. Y. P.; Akbar, A. Trends Pharm. Sci. 2015, 36, 32–40.
- Keillor, J. W. In Transglutaminases: Multiple Functional Modifiers and Targets for New Drug Discovery; Hitomi, K.; Kojima, S.; Fésüs, L., Eds.; Transglutaminases: Multiple Functional Modifiers and Targets for New Drug Discovery; Springer Japan: Tokyo, 2015; pp. 347–372.
- Keillor, J. W.; Apperley, K. Y. P. Expert Opin. Ther. Pat. 2016, 26, 49–63.
- Marrano, C.; de Macédo, P.; Keillor, J. W. Bioorg. Med. Chem. 2001, 9, 1923–1928.
- Marrano, C.; de Macédo, P.; Gagnon, P.; Lapierre, D.; Gravel, C.; Keillor, J. W. Bioorg. Med. Chem. 2001, 9, 3231–3241.
- Halim, D.; Caron, K.; Keillor, J. W. Bioorg. Med. Chem. Lett. 2007, 17, 305–308.
- Pardin, C.; Gillet, S. M. F. G.; Keillor, J. W. Bioorg. Med. Chem. 2006, 14, 8379–8385.
- Keillor, J. W.; Chica, R. A.; Chabot, N.; Vinci, V.; Pardin, C.; Fortin, E.; Gillet, S. M. F. G.; Nakano, Y.; Kaartinen, M. T.; Pelletier, J. N.; Lubell, W. D. Can. J. Chem. 2008, 86, 271–276.
- Al-Jallad, H. F.; Myneni, V. D.; Piercy-Kotb, S. A.; Chabot, N.; Mulani, A.; Keillor, J. W.; Kaartinen, M. T. PLoS ONE 2011, 6, e15893.
- Song, Y.; Kirkpatrick, L. L.; Schilling, A. B.; Helseth, D. L.; Chabot, N.; Keillor, J. W.; Johnson, G. V. W.; Brady, S. T. Neuron 2013, 78, 109–123.
- Chabot, N.; Moreau, S.; Mulani, A.; Moreau, P.; Keillor, J. W. Chem. Biol. 2010, 17, 1143–1150.
- Clouthier, C. M.; Mironov, G. G.; Okhonin, V.; Berezovski, M. V.; Keillor, J. W. Angew. Chem. Int. Ed. Engl. 2012, 51, 12464–12468.
- Akbar, A.; Keillor, J. W. unpublished.
- Pinkas, D. M.; Strop, P.; Brunger, A. T.; Khosla, C. PLoS Biol. 2007, 5, e327.
- Fisher, M. L.; Keillor, J. W.; Xu, W.; Eckert, R. L.; Kerr, C. Mol. Cancer Res. 2015, 13, 1083–1094.
- Fisher, M. L.; Adhikary, G.; Xu, W.; Kerr, C.; Keillor, J. W.; Eckert, R. L. Oncotarget 2015, 6, 20525–20539.
The author:
Jeffrey Keillor is a full professor in the department of Chemistry and Biomolecular Sciences at the University of Ottawa, where he holds a University Research Chair in Bioorganic Chemistry. In addition to developing new methods for site-specific protein labelling, he has studied the mechanism and inhibition of transglutaminases since 1995. His most recent collaborative work has focussed on the inhibition of TG2 in cancer cells.
 Besuch des Bundesministers für Wirtschaft und Klimaschutz Dr. Robert Habeck bei der Zedira
Besuch des Bundesministers für Wirtschaft und Klimaschutz Dr. Robert Habeck bei der Zedira  Discover Our New Catalogue Edition and Dive into the World of Transglutaminases!
Discover Our New Catalogue Edition and Dive into the World of Transglutaminases!  Successful ISO9001:2015 recertification
Successful ISO9001:2015 recertification  Dr. Falk Pharma and Zedira announce successful completion of the phase 2a proof-of-concept study of ZED1227 for the treatment of Celiac Disease
Dr. Falk Pharma and Zedira announce successful completion of the phase 2a proof-of-concept study of ZED1227 for the treatment of Celiac Disease  Dr. Falk Pharma und Zedira verkünden den erfolgreichen Abschluss der Phase 2a-Studie mit ZED1227 zur Behandlung von Zöliakie
Dr. Falk Pharma und Zedira verkünden den erfolgreichen Abschluss der Phase 2a-Studie mit ZED1227 zur Behandlung von Zöliakie  Reversibly acting transglutaminase 2 inhibitors: drug candidates for the treatment of fibrosis
Reversibly acting transglutaminase 2 inhibitors: drug candidates for the treatment of fibrosis  Transcriptomic analysis of the efficacy of TG2-inhibitor trials and human intestinal organoids modelling Celiac disease pathogenesis
Transcriptomic analysis of the efficacy of TG2-inhibitor trials and human intestinal organoids modelling Celiac disease pathogenesis  Transglutaminase antibodies and neurological manifestations of gluten sensitivity
Transglutaminase antibodies and neurological manifestations of gluten sensitivity  Design of Oral FXIIIa Blockers as Safer Anticoagulants Mission Impossible?
Design of Oral FXIIIa Blockers as Safer Anticoagulants Mission Impossible?  Microbial transglutaminase (MTG) enables efficient and site-specific conjugation to native antibodies without the need of antibody engineering
Microbial transglutaminase (MTG) enables efficient and site-specific conjugation to native antibodies without the need of antibody engineering  Tridegin as FXIIIa inhibitor
Tridegin as FXIIIa inhibitor  Microbial transglutaminase: from discovery to market
Microbial transglutaminase: from discovery to market  Tissue transglutaminase inhibitors
Tissue transglutaminase inhibitors  Tissue transglutaminase in Alzheimers Disease
Tissue transglutaminase in Alzheimers Disease  Factor XIIIa: novel target for anticoagulation?
Factor XIIIa: novel target for anticoagulation?  Microbial transglutaminase for site-specific protein conjugation
Microbial transglutaminase for site-specific protein conjugation